IonQuant is a fast and comprehensive tool for MS1 precursor intensity-based quantification for timsTOF PASEF DDA and non-timsTOF (e.g., Orbitrap) data. It enables label-free quantification with false discovery (FDR) controlled match-between-runs (MBR). It can also be used for quantification in labelling-based experiments such as those involving SILAC, dimethyl, or similar labelling strategies. IonQuant is available as a stand-alone tool or it can be run within FragPipe computational platform.
Workflow of peak tracing and quantification

Workflow of FDR-controlled MBR

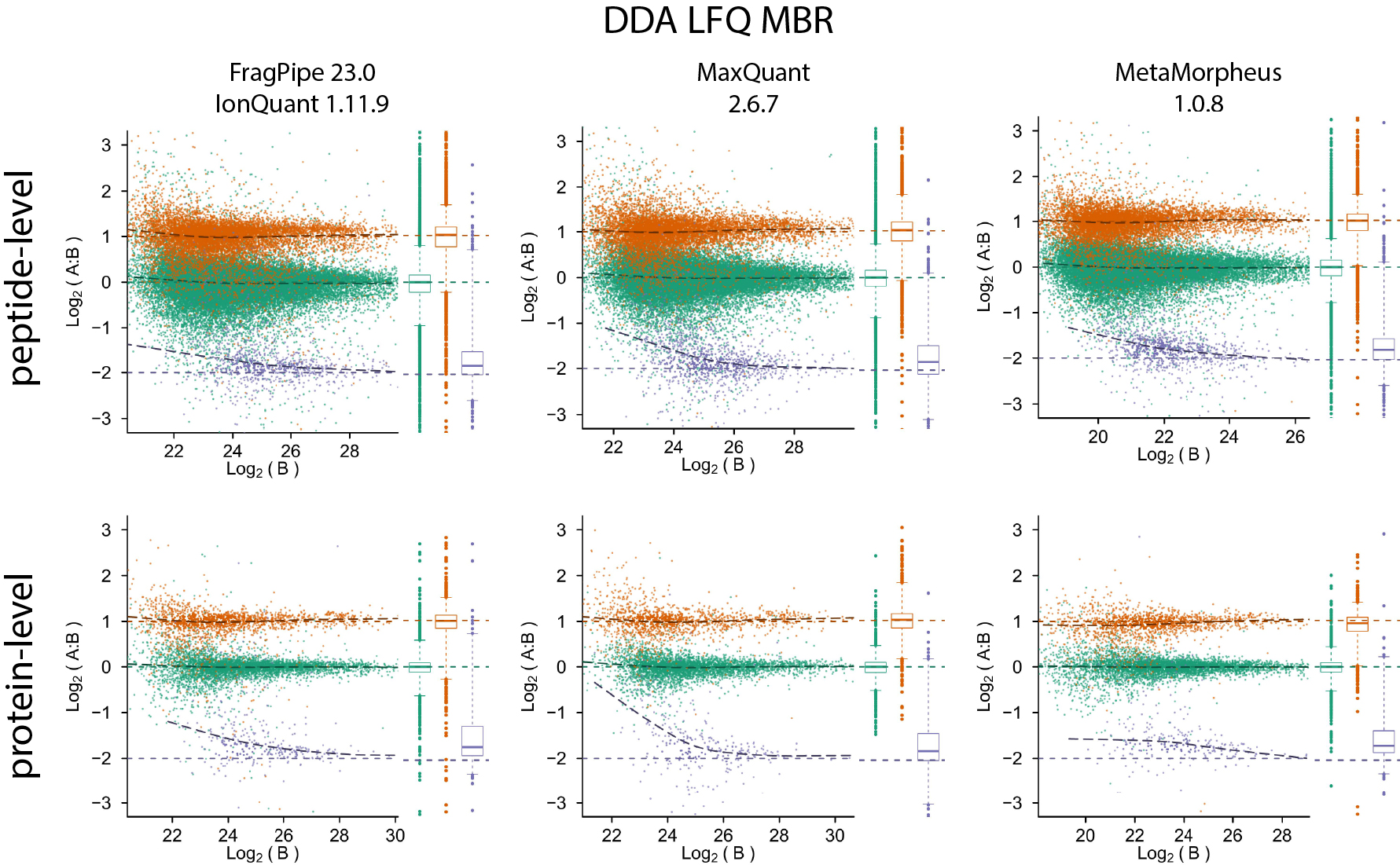
It is accurate and sensitive
Quantification accuracy and precision evaluation using the PXD003881 (IonStar) dataset
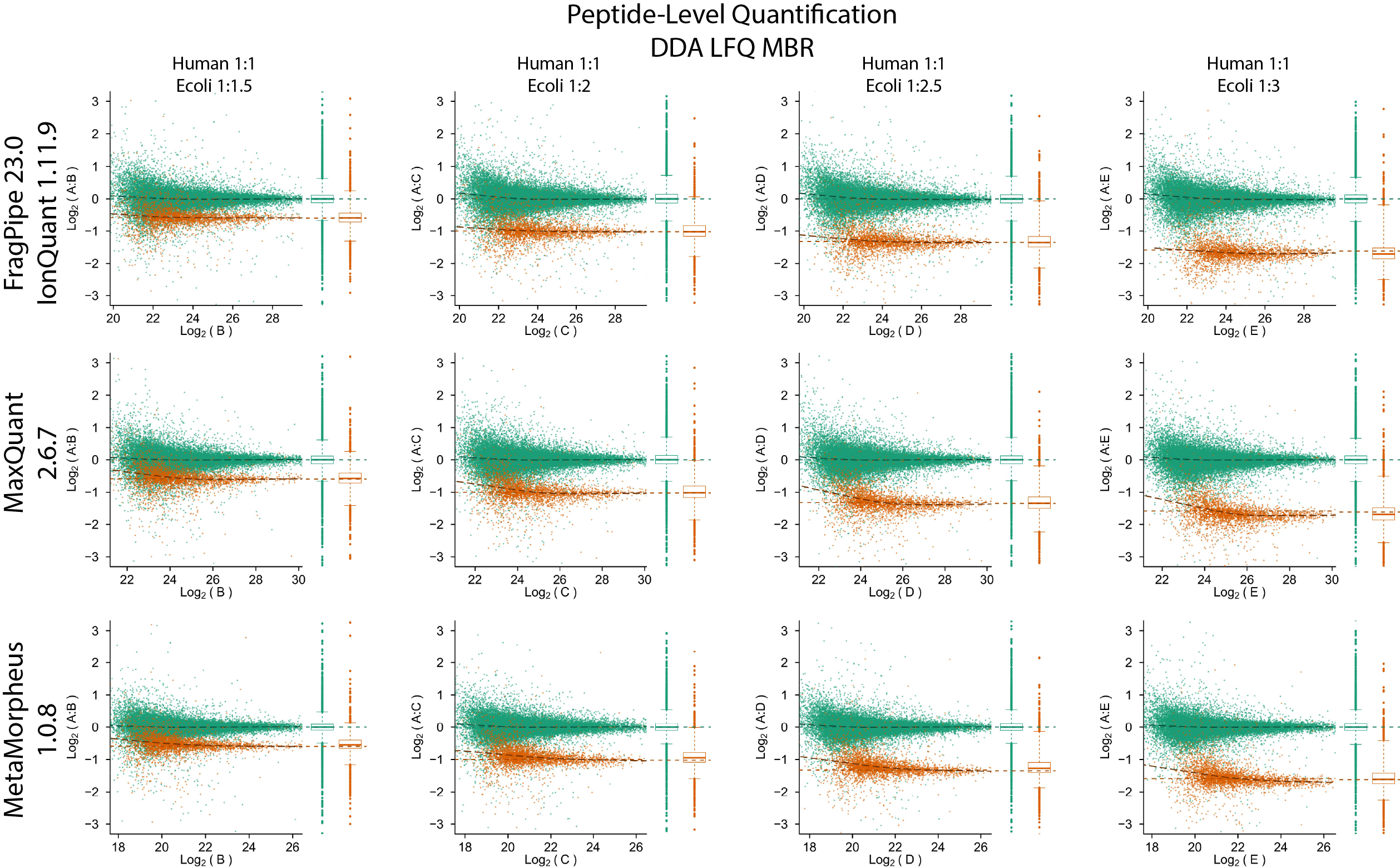
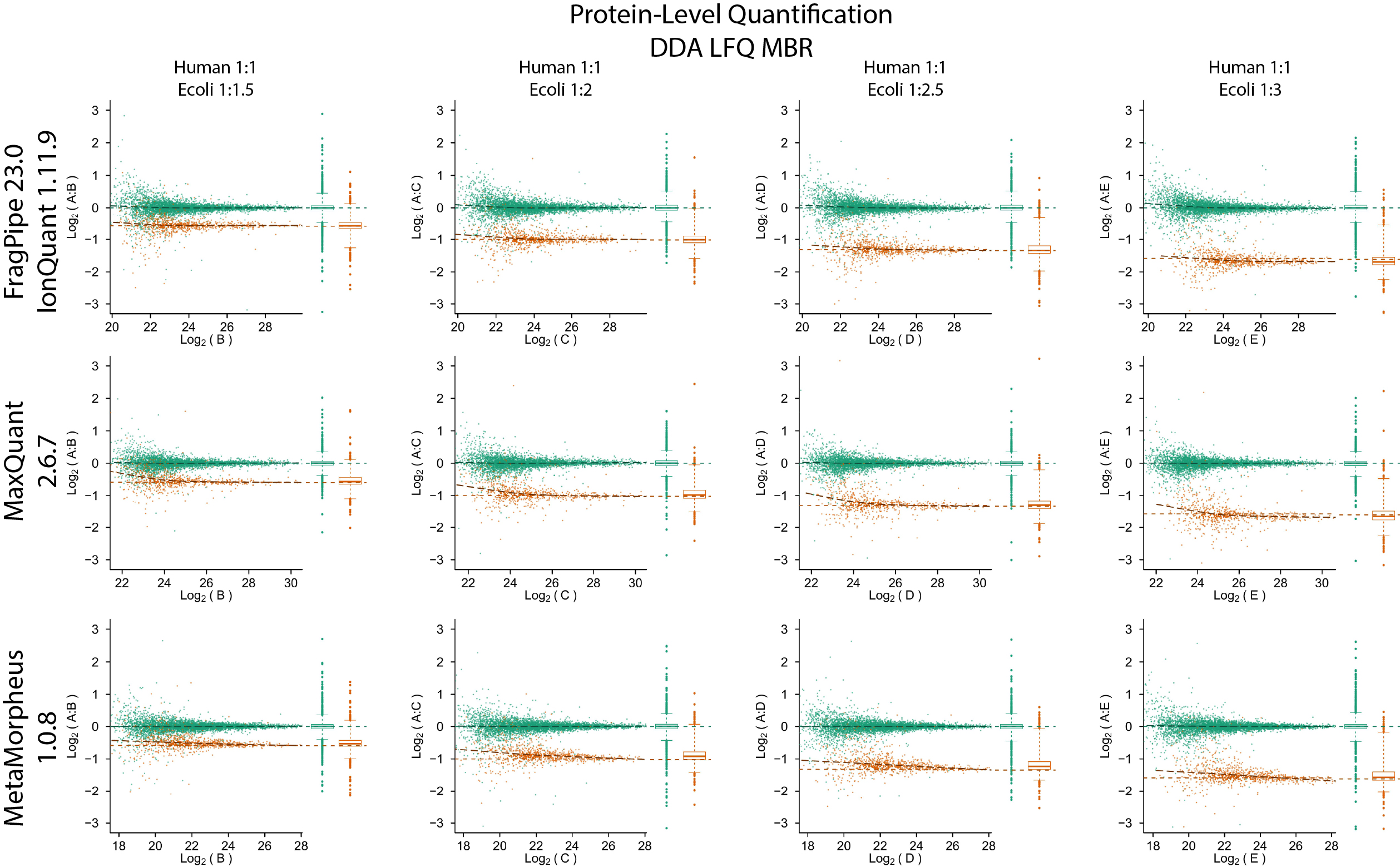
Quantification accuracy and precision evaluation using the PXD028735 dataset
It is fast

System requirements
- Java 11+.
extfolder from the latest MSFragger.- The latest FragPipe from here (optional).
Note: Bruker’s native library needs Visual C++ Redistributable for Visual Studio 2017 in Windows. If you see an error saying cannot find Bruker native library, please try to install the Visual C++ redistibutable.
License
IonQuant is available freely for academic research, non-commercial or educational purposes under academic license.
Other uses require a commercial license that can be obtained by visiting Fragmatics or emailing at info@fragmatics.com.
Download
Academic users can downloaded IonQuant from here. Comercial users must contact Fragmatics to obtain the executable of the program.
Changelog
Changelog can be found from here.
Usage
Users can also run IonQuant standalone in command line interface
- Download the standalone zip file from here.
- Run
java -jar IonQuant.jarto print the help document.
Usage:
java -jar IonQuant.jar <options> --specdir <one directory to the spectral files> --psm <path to psm.tsv file> --psm <path to psm.tsv file>...
OR
java -jar IonQuant.jar <options> --filelist <path to filelist file>
Options:
--specdir <string> # Directory containing the spectral files (d/mzml/mzxml/raw/quantindex). One --specdir indicates one spectral directory and can have multiple --specdir.
--perform-ms1quant 0/1 # Perform MS1 quantification. 0 = no, 1 = yes. Default: 1
--perform-isoquant 0/1 # Perform isobaric labeling quantification. 0 = no, 1 = yes. Default: 0
--isotol <float> # MS2 tolerance in ppm. Default: 10
--isolevel <int> # Isobaric quantification level. 2 = MS2, 3 = MS3, 4 = ZOOM-HR. Default: 2
--isotype <string> # Isobaric quantification type. Case insensitive. Support iTRAQ-4, iTRAQ-8, TMT-0, TMT-2, TMT-6, TMT-10, TMT-11, TMT-16, TMT-18, TMT-35, sCLIP-6, iBT-16, DiLeu-12, DiLeu-1, DeAla-13, iodoTMT-6. Default: TMT-10
--annotation <string> # Annotation file info for the isobaric quantification. Format: <path to psm.tsv>=<path to annotation file>. One --annotation indicates one annotation file and can have multiple --annotation. Default: <blank>
--site-reports 0/1 # Generate site reports. 0 = no, 1 = yes. The psm.tsv need to have the modification localization modification columns. Default: 1
--msstats 0/1 # Generate MSstats input files. 0 = no, 1 = yes. Default: 0
--threads <integer> # Number of threads. 0 = all logical cores. Default: 0
--mztol <float> # MS1 tolerance in PPM. Default: 10.0
--imtol <float> # 1/K0 tolerance. Default: 0.05
--rttol <float> # Retention time tolerance. Unit: min. Default: 0.4
--psm <string> # Path to Philosopher's psm.tsv. One --psm indicates one psm.tsv and can have multiple --psm.
--multidir <string> # Output directory for the multi-experimental result. Optional. Default: <blank>
--normalization 0/1 # Normalize the intensities across all runs. Default: 1
--minisotopes 1/2/3 # Minimum isotopes required in feature extraction. Default: 2
--minscans <integer> # Minimum MS1 scans required in feature extraction. Default: 3
--minions <integer> # Minimum ions required in quantifying proteins. Only for MaxLFQ intensity. Default: 1
--excludemods <string> # Excluded modifications in quantifying peptide sequences and proteins. Format: <amino acid><mass>;... Default: <blank>
--maxlfq 0/1 # Calculate MaxLFQ intensity. 0 = no, 1 = yes. Default: 1
--ibaq 0/1 # [experimental] Calculate iBAQ intensity. The iBAQ intensity is normalized by the protein length, not the number of theoretical peptides. 0 = no, 1 = yes. Default: 0
--minexps <int> # Minimum experiments in picking an ion for quantifying proteins. Only for intensity, not for MaxLFQ intensity. Default: 1
--minfreq <float> # Minimum required frequency of an ion being selected for protein quantification. Only for intensity, not for MaxLFQ intensity. Default: 0
--tp <int> # Number of ions used in quantifying each protein. If 0, using all ions. For intensity, not for MaxLFQ intensity. Default: 0
--mbr 0/1 # Perform match-between-runs. Default: 0
--mbrrttol <float> # Retention time tolerance used in match-between-runs. Unit: min. Default: 1.0
--mbrimtol <float> # 1/K0 tolerance used in match-between-runs. Default: 0.05
--mbrtoprun <integer> # Maximum number of donor runs for each acceptor run. Default: 10
--mbrmincorr <float> # Minimum correlation coefficient between a donor run and its acceptor run. Default: 0
--ionmobility 0/1 # The data has ion mobility information or not (for conventional LC-MS data). Default: 0
--ionfdr <float> # Transferred ion false discovery rate threshold. Default: 0.01
--peptidefdr <float> # Transferred peptide false discovery rate threshold. Default: 1
--proteinfdr <float> # Transferred protein false discovery rate threshold. Default: 1
--light <string> # Light labelling mass. Format: <amino acids><mass>;<amino acids><mass>;... Optional. Default: <blank>
--medium <string> # Medium labelling mass. Format: <amino acids><mass>;<amino acids><mass>;... Optional. Default: <blank>
--heavy <string> # Heavy labelling mass. Format: <amino acids><mass>;<amino acids><mass>;... Optional. Default: <blank>
--formula <string> # List the formula of the modifications used in the data. Required if want to perform isotope-labeling quantification. Format: <chemical composition>;<chemical composition>... example: C(2)H(2)O;HO(3)P;2H(4)C(2). Default: <blank>
--requantify 0/1 # Re-quantify unidentified feature based on identified feature. Only activate when --light, --medium, or --heavy is not empty. Default: 1
--writeindex 0/1 # Write indexed file on disk for further usage. 0 = no, 1 = yes. Default: 0
--locprob <float> # Localization probability threshold. Default: 0
--filelist <string> # A file containing flags. Default: <blank>
--uniqueness 0/1/2 # Peptide-protein uniqueness. 0 = unique+razor, 1 = unique only, 2 = all. Default: 0
--intensitymode 0/1/2 # The mode to calculate the ion intensity. 0 = apex, 1 = area, 2 = auto. Default: 2
--modlist <string> # A file lists modification masses. Those masses are used to remove the mass discrepancy due to rounding errors. Default: <blank>
Tutorials:
- Using FragPipe coupled with MSFragger and IonQuant to analyze samples
- Analyzing SILAC (or other labelling coupled with MS1-based quantification) samples with FragPipe
- Running MSstats using MSstats.csv from IonQuant
Publications
Fast quantitative analysis of timsTOF PASEF data with MSFragger and IonQuant
Fengchao Yu, Sarah E. Haynes, Guo Ci Teo, Dmitry M. Avtonomov, Daniel A. Polasky, Alexey I. Nesvizhskii
Molecular & Cellular Proteomics, 19 (2020), 1575-1585, DOI: 10.1074/mcp.TIR120.002048
IonQuant Enables Accurate and Sensitive Label-Free Quantification With FDR-Controlled Match-Between-Runs
Fengchao Yu, Sarah E. Haynes, Alexey I. Nesvizhskii
Molecular & Cellular Proteomics, 20 (2021), 100077, DOI: 10.1016/j.mcpro.2021.100077